10 Changes to the FDA’s Pre-Cert Program Model
In June 2018 the FDA released an update to its working model of the Software Precertification...
Read More

In June 2018 the FDA released an update to its working model of the Software Precertification...
Read More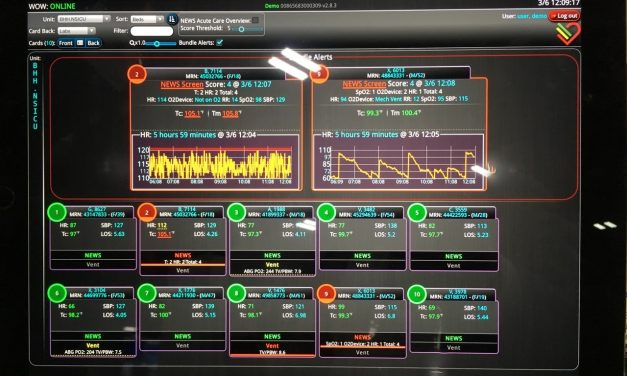
In the May 10th Pre-Cert webinar the FDA addressed an interesting risk matrix that was previously...
Read More
The impact of FDA's digital health Pre-Cert program has yet to be fully seen. There is much debate...
Read More
Since connectivity runs on software, and some medically related software is a medical device, and...
Read More
On October 25, 2017 the FDA released its guidance on "Deciding When to Submit a 510(k) for a...
Read More
The FDA, medical devices, and cybersecurity are popular subject matter for proposed Federal...
Read More
Cybersecurity continues to be a hot topic in healthcare with several areas of concern. These...
Read More
Software as a Medical Device (SaMD) is terminology under the aegis of a work group of...
Read More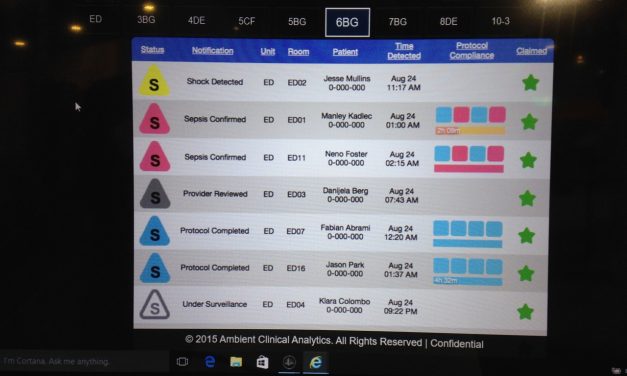
A ubiquitous characteristic of software is that it often undergoes numerous changes after it is...
Read More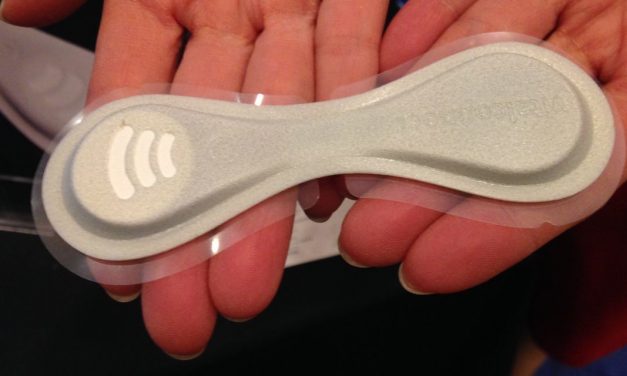
Among the many forms of data flow that might occur from a medical device is direct to the patient....
Read More
Recent Comments