July 2019 SaMD PreCert Update
We have been following the FDA's Software as Medical Device (SaMD) Pre-Cert pilot. The premise of...
Read More

We have been following the FDA's Software as Medical Device (SaMD) Pre-Cert pilot. The premise of...
Read More
Some of us remember when distributed medical devices such as patient monitors had their own...
Read More
FDA has issued a request for SaMD developers to try out the PreCert excellence appraisal process....
Read More
Recently the FDA released a discussion paper on a Proposed Regulatory Framework for Modifications...
Read MoreWe have addressed the FDA Pre-Cert program for SaMD (Software as Medical Device) on several...
Read More
The FDA has released a January 2019 update to its working model of the precertification...
Read More
The 21st Century Cures Act of 2016 (Cures) had many provisions including defining what kinds of...
Read More
Medical devices that contain software, or are software, are subject to the well-known medical...
Read More
Uncertainty abounds when managing digital health regulatory uncertainty regarding the FDA and...
Read More
The FDA has released its plans for Final and Draft Guidance Documents for fiscal year 2019. ...
Read More
The Office of the Inspector General (OIG) of Health and Human Services (HHS) recently released a...
Read More
In June 2018 the FDA released an update to its working model of the Software Precertification...
Read More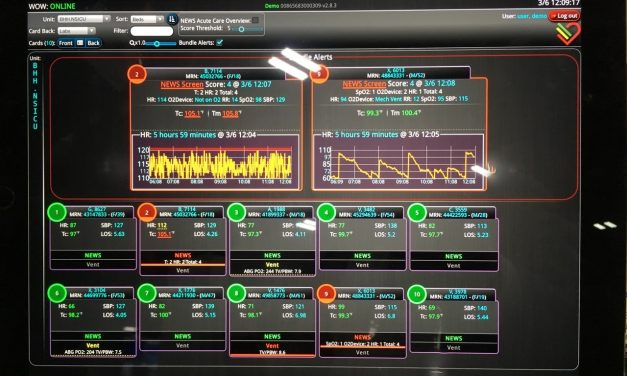
In the May 10th Pre-Cert webinar the FDA addressed an interesting risk matrix that was previously...
Read More
The impact of FDA's digital health Pre-Cert program has yet to be fully seen. There is much debate...
Read More
Since connectivity runs on software, and some medically related software is a medical device, and...
Read More

Recent Comments